Command Line¶
A large subset of ontobio functionality is available via a powerful command line interface that can be used by non-programmers.
You will first need to install, see Installation
After that, set up your PATH:
export PATH $HOME/repos/ontobio/ontobio/bin
ogr -h
For many operations you need to be connected to a network
Note: command line interface may change
Ontologies¶
The ogr command handles ontologies
Connecting to ontologies¶
Specify an ontology with the -r option. this will always be the OBO
name, for example go, cl, mp, etc
-r goconnect to GO via default method (currently OntoBee-SPARQL)-r obo:goconnect to GO via download and cache of ontology from OBO Library PURL-r /users/my/my-ontologies/go.jsonuse local download of ontology
See Inputs for possible sources to connect to
In the following we assume default method, but the -r argument can be substituted.
Basic queries¶
Show all classes named neuron:
ogr -r cl neuron
Multiple arguments can be provided, e.g.:
ogr -r cl neuron hepatocyte erythrocyte
Ancestors queries¶
List all ancestors:
ogr -r cl neuron
Show ancestors as tree, following only subclass:
ogr -r cl -p subClassOf -t tree neuron
generates:
% GO:0005623 ! cell
% CL:0000003 ! native cell
% CL:0000255 ! eukaryotic cell
% CL:0000548 ! animal cell
% CL:0002319 ! neural cell
% CL:0000540 ! neuron *
% CL:0002371 ! somatic cell
% CL:0002319 ! neural cell
% CL:0000540 ! neuron *
Descendants of neuron, parts and subtypes
ogr -r cl -p subClassOf -p BFO:0000050 -t tree -d d neuron
Descendants and ancestors of neuron, parts and subtypes
ogr -r cl -p subClassOf -p BFO:0000050 -t tree -d du neuron
All ancestors of all classes 2 levels down from subclass-roots within CL:
ogr -r cl -P CL -p subClassOf -t tree -d u -L 2
Visualization using obographviz¶
Requires: https://www.npmjs.com/package/obographviz
Add og2dot.js to path
ogr -p subClassOf BFO:0000050 -r go -t png a nucleus
This proceeds by:
- Using the python ontobio library to extract a networkx subgraph around the specified node
- Write as obographs-json
- Calls og2dot.js
Output:
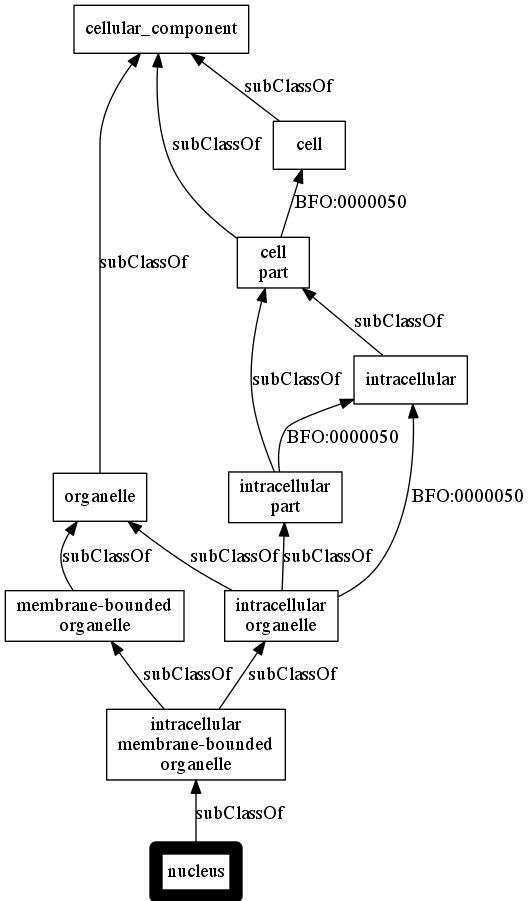
img
Search¶
List exact matches to neuron
ogr -r cl neuron
Terms starting with neuron, SQL style
ogr -r cl neuron%
Terms starting with neuron, regex (equivalent to above)
ogr -r cl -s r ^neuron
Terms ending with neuron
ogr -r cl -s r neuron$
Terms containing the string neuron
ogr -r cl -s r neuron
Note: any of the above can be fed into other renderers, e.g. trees, graphs
E.g. terms containing neuron
ogr -r cl %neuron%
E.g. terms ending neuron, to tree
ogr -r cl %neuron -t tree
Properties¶
Properties (relations) are treated as nodes in the graph, e.g.
ogr-tree -d ud -r ro 'develops from'
. RO:0002324 ! developmentally related to
% RO:0002258 ! developmentally preceded by
% RO:0002202 ! develops from *
% RO:0002225 ! develops from part of
% RO:0002494 ! transformation of
% RO:0002495 ! immediate transformation of
% RO:0002207 ! directly develops from
% RO:0002495 ! immediate transformation of
SPARQL integration¶
SPARQL where clauses can be inserted using -Q to pipe the results
of a query to generate the initial set of IDs, e.g.:
ogr-tree -r pato -Q "{?x rdfs:subClassOf+ PATO:0000052}"
Associations¶
The ontobio-assoc command handles ontologies
Subcommands:
subontology Extract sub-ontology
enrichment Perform an enrichment test
phenolog Perform multiple enrichment tests
query Query based on positive and negative terms
associations Query for association pairs
intersections Query intersections
dendrogram Plot dendrogram from intersections
simmatrix Plot dendrogram for similarities between subjects
Examples¶
Enrichment analysis, using all genes associated to a GO term as sample (we expect this GO term to be top results)
ontobio-assoc -v -r go -T NCBITaxon:9606 -C gene function enrichment -q GO:1903010
Plotly:
ontobio-assoc -v -r go -T NCBITaxon:10090 -C gene function dendrogram GO:0003700 GO:0005215 GO:0005634 GO:0005737 GO:0005739 GO:0005694 GO:0005730 GO:0000228 GO:0000262
Show similarity matrix for a set of genes:
ontobio-assoc -v -r go -T NCBITaxon:10090 -C gene function simmatrix MGI:1890081 MGI:97487 MGI:106593 MGI:97250 MGI:2151057 MGI:1347473
Basic queries, using file as input:
ontobio-assoc -C gene function -T pombe -r go -f tests/resources/truncated-pombase.gaf query -q GO:0005622
Parsing assoc files¶
The ontobio-parse-assocs.py command will parse, validate and
convert association files (GAF, GPAD, HPOA etc) of all file types and versions.
Top Level Options¶
ontobio-parse-assocs.py mostly uses top level options before subcommands to configure parsing.
-r, --resourceis the ontology file, in OBO JSON format-f, --fileinput annotation file-F, --formatis the format of the input file. GAF will be the default if not provided--report-mdand--report-jsonare the paths to output the parsing and validation reports to
Use validate to produce a report validating the input file, -f, --file.
Use convert to convert the input annotation file into a GPAD or GAF of any version. A report will still be produced.
* -t, --to is the format to convert to. GAF, GPAD are accepted.
* -n, --format-version is the version. For GAF, 2.1 or 2.2 are accepted with 1.2 as default. For GPAD 1.2 or 2.0 are accepted with 1.2 default.
GO Rules¶
ontobio-parse-assocs.py is capable of running the GO Rules (https://github.com/geneontology/go-site/tree/master/metadata/rules) over each annotation as they are parsed. By default, in this script, annotations are not validated by GO Rules except gorule-0000020, gorule-0000027, and gorule-0000059.
To include a rule in the rule set use the option -l or --rule followed by an integer representing the rule ID.
For example to include gorule-0000006:
ontobio-parse-assocs.py -f my_assoc.gaf --report-md report.md -l 6 validate
Use multiple -l <ID> to build up a list of rules that will be used to validate the input file:
ontobio-parse-assocs.py -f my_assoc.gaf --report-md report.md -l 6 -l 13 validate
To turn on all rules at once, use -l all:
ontobio-parse-assocs.py -f my_assoc.gaf --report-md report.md -l all validate
Under the hood, this is all controlled using a parameter, rule_set attached to the AssocParserConfig class. This accepts a list of integers or the string "all" or None. Setting to None (the default) will include no rules, and using "all" will use all rules.
The parameter passed in is used to create the assocparser.RuleSet dataclass.
GOlr Queries¶
The qbiogolr.py command is for querying a GOlr instance